A neural journey into disease transmission models
Let’s start with an elementary, but yet insightful equation system commonly known as the SIS disease transmission model. Full stop. Let’s start from somewhere else.
The fact that biological entitities such as viruses, bacteria and fungi have durably colonized the human body sets the stage for a life-long antagonistic relationship between them and us - the parasites and the hosts. But not only this unfriendly relationship sets them apart from each other, these best enemies live on distinct spatial and temporal scales. Viruses are sized in the micrometers and can only be observed via electronic microscopy, and one virus can produce offspring in the tens of thousands within hours or days. On the other hand, multicellular tissue heaps known as humans are sized in meters, and it takes decades for them to reproduce, if at all. The action starts when parasites enter human cells, mainly by tricking their surface proteins and making it into the cellular reproduction machinery. Parasites exploit the host cell for their own metabolism or reproduction. Eventually the host cell will disintegrate, parasite offspring is released into the cellular environment to find its next victim. But one host is not enough. Insatiable as they are, the parasites need to transmit in order to continue living, as their fate is bound to the host. In order to enter into new hosts, parasites undergo continuous selection, their genetic makeup changes randomly as they replicate. Full stop.
For microbiologists, this description of a parasite’s lifecycle might sound like an overly simplified vulgarisation, yet it is a physicist’s nightmare: biochemical entities interacting at multiple scales. From the medical perspective, we are dealing with discrete, host-centric events: life, death, infection, symptoms, recovery. This is where disease transmission models start from. Disregarding the complexities of biochemical interactions that allow the parasites to enter host cells and ignoring the evolutionary ecology of parasite population dynamics, they assign each host to a compartment. Like in the old days, when train coachs were divided into compartments, one could comfortably talk, eat and smoke (sic!) in the compartment, granted that all individuals in the compartment would be alike. In the same way, all hosts that share attributes such as susceptible (to infection), infectious (to other hosts) or recovered (from infection) are put into their respective compartment. A rather ad-hoc set of rules defines how hosts can change between compartments, e.g. an infectious host might recover after some time, in the same way as people move from the non-smoking to the smoking compartment on the train. Disease transmission models track the size of the compartments over time. Differential equation techniques of the ordinary, stochastic, delay or partial kind have been invoked to produce model solution curves to be aligned with epidemiological observations.
Let’s start again with an elementary system of differential equations, which models the temporal evoluation of only two compartments, susceptible (S) and infectious hosts (I). Let’s denote the total population size with \(N=S+I\) and assume that \(N\) remains constant. We posit two ad-hoc rules of transitioning between compartments.
First, for each susceptible host, the probability of finding an infectious host in the population is \(\tfrac{I}{N}\), and since there are \(S\) susceptible hosts, we would have on average \(\beta S \tfrac{I}{N}\) susceptible hosts moving to the (I) compartment. The parameter \(\beta>0\) encapsulates all the biochemical and eco-evolutionary complexities that we proudly ignored.
Second, infectious hosts might also recover from infection and move to the susceptible compartment with rate \(\gamma\), and we would end up with \(\gamma I\) hosts on overage moving from the (I) compartment to the (S) compartment.
Differential equations bear their name from the physical principle of motion. In order to describe the trajectory of an object, we only need to know its initial position and its velocity. From the point of view of measurement, velocity should be seen as change of position between two consecutive time points. Mathematicians would paraphrase this measurement of change as difference quotient, and as the distance between consecutive time points gets very small, they would refer to the change as differential quotient or short differential. In its simplest form, this differential depends only on the position itself. So, differential equation, here you are! On your left-hand side, we put the differential to measure infinitesimal change in position, on your right-hand side we put some function of position and other things to quantify the strength of change. Mathematicians are fond of definitions, and even have a name for your right-hand side: they call it a vector field.
Our SIS system reads:
\[\begin{aligned} \label{eq:sis} \frac{dS}{dt} &= -\beta S \tfrac{I}{N} + \gamma I \\ \frac{dI}{dt} &= \beta S \tfrac{I}{N} - \gamma I \end{aligned}\]Under the assumptions of constant population size we set \(S=N-I\) and the SIS system simplifies to a single equation: \begin{equation} \label{eq:sis_simple} \frac{dI}{dt} = \beta (N-I) \tfrac{I}{N} - \gamma I = (\beta - \gamma)I- \tfrac{\beta}{N}I^2 \end{equation}
Our differential is \(\frac{dI}{dt}\) and the vector field reads \(f(x)=(\beta - \gamma) x - \tfrac{\beta}{N}x^2\). The linear part \(x\mapsto (\beta - \gamma)x\) of the vector field describes how the balance between new infections and recovery relates to the growth of (I), especially in the beginning. If \(\beta - \gamma<0\), negative growth will push eventually the system to the state \(I=0\), the so-called disease-free equilibrium. If \(\beta - \gamma>0\), the non-linear part \(x\mapsto -\tfrac{\beta}{N}x^2\) of the vector field helps to keep the system in place: there cannot be more than \(N\) hosts! If \(I\) is close to \(N\), the \(\beta / N\) terms cancel out and \(x\mapsto -\gamma x\) will push the sytem to lower values of \(I\), it will eventually reach \(I=N\tfrac{\beta - \gamma}{\beta}=N\left(1-\tfrac{\gamma}{\beta}\right)\), the so-called endemic equilibrium.
To solve the differential equation $\frac{dI}{dt} = f(I)$ we evaluate the vector field at discrete, equidistant time points $t_i=t_0+i\Delta$:
$\begin{equation} \label{eq:sis_simple_num} \frac{I(t_{i+1})-I(t_i)}{\Delta} = f(I(t_i)) \end{equation}$ and rewrite the equation as a recurrence relationship $I(t_{i+1})=I(t_i)+\Delta f(I(t_i))$. If you know the initial condition $I(t_0)$, then you can calculate $I(t_1), I(t_2),\dots$
This recurrence relationship is also known as (forward) Euler schema. It is the first among many numerical methods to solve differential equations. In the machine learning community, similar algorithms have been utilized under the name of residual neural networks. Again, we have “information blocks” (e.g. vectors or tensors of real numbers) that are indexed with $i, i+1,\dots$. In order to move from the \(i\) th block to the \((i+1)\) th block, a neural network element $\mathfrak{f}$ (e.g. a vector-valued function) is used. Here, $\mathfrak{f}$ could be any continuous mapping that takes a vector of size $n$ into a vector of size $m$. Often, neural network elements use elementary functions such as step functions or sigmoids and combine them by algebraic operations. You can think of combining resistors, capacitators and switches in your electric circuit board to form a electric network. In the same way as resistors in your electric circuit control the electric current, every operation between two elementary functions carries a weight. One can even switch off entire parts the network, or keep the weights at a very low level. Like capacitators that can store and release electric energy more or less quickly, the elements of neural networks can be tuned to smooth the outgoing signal. In machine learning this is called hyperparameter tuning. The machine learning task is to infer the weights such that the electric energy that you put into your circuit reproduces the flinkering LED as the output of your particular circuit.
Let us think of our differential equation model as a simple, serial electrical circuit. We had an explicit form for $\mathfrak{f}$. Seen as a neural network element, it takes a real number and produces another real number with unknown weights $w=(w_1, w_2, w_3)$:
\[\mathfrak{f}_w: x \mapsto w_1 x \oplus w_2 x \oplus (w_3 x\otimes x)\]This neural network element \(\mathfrak{f}\) should look very familiar. If we set $w_1=\beta, w_2=-\gamma, w_3=-\tfrac{\beta}{N}$, it is exactly our vector field \(f(x)=(\beta - \gamma) x - \tfrac{\beta}{N}x^2\). The operators $\otimes$ resp. $\oplus$ are called tensor product resp. direct sum.
Residual neural networks (ResNets) translate the idea of Euler schema to neural networks:
\[I(t_{i+1})=I(t_i) \oplus \mathfrak{f}(I(t_i))\]In order to move to the next block, you simple take the value of your current block and update it in the direction of the neural network element.
We have invistigated our differential equation with vector field and neural network glasses. But there is yet another way of looking at it. We can also integrate the differential equation. By definition of the integral, we obtain $I(t_{i+1})-I(t_i)= \int_{t_i}^{t_{i+1}} f(I(t)) dt$ and for small $\Delta>0$ we can assume $\int_{t_i}^{t_{i+1}} f(I(t)) dt\sim \Delta f(I(t_i)) $. We solve our differential equation by literally integrating along the vector field $f$. For this reason $t\mapsto I(t)$ is also called an integral curve. By definition, the vector field is tangent to the integral curve. If we know the tangent directions and strength for every state of our system, we can construct integral curves. If we now integral curves from every possible initial condition, we can construct vector fields.
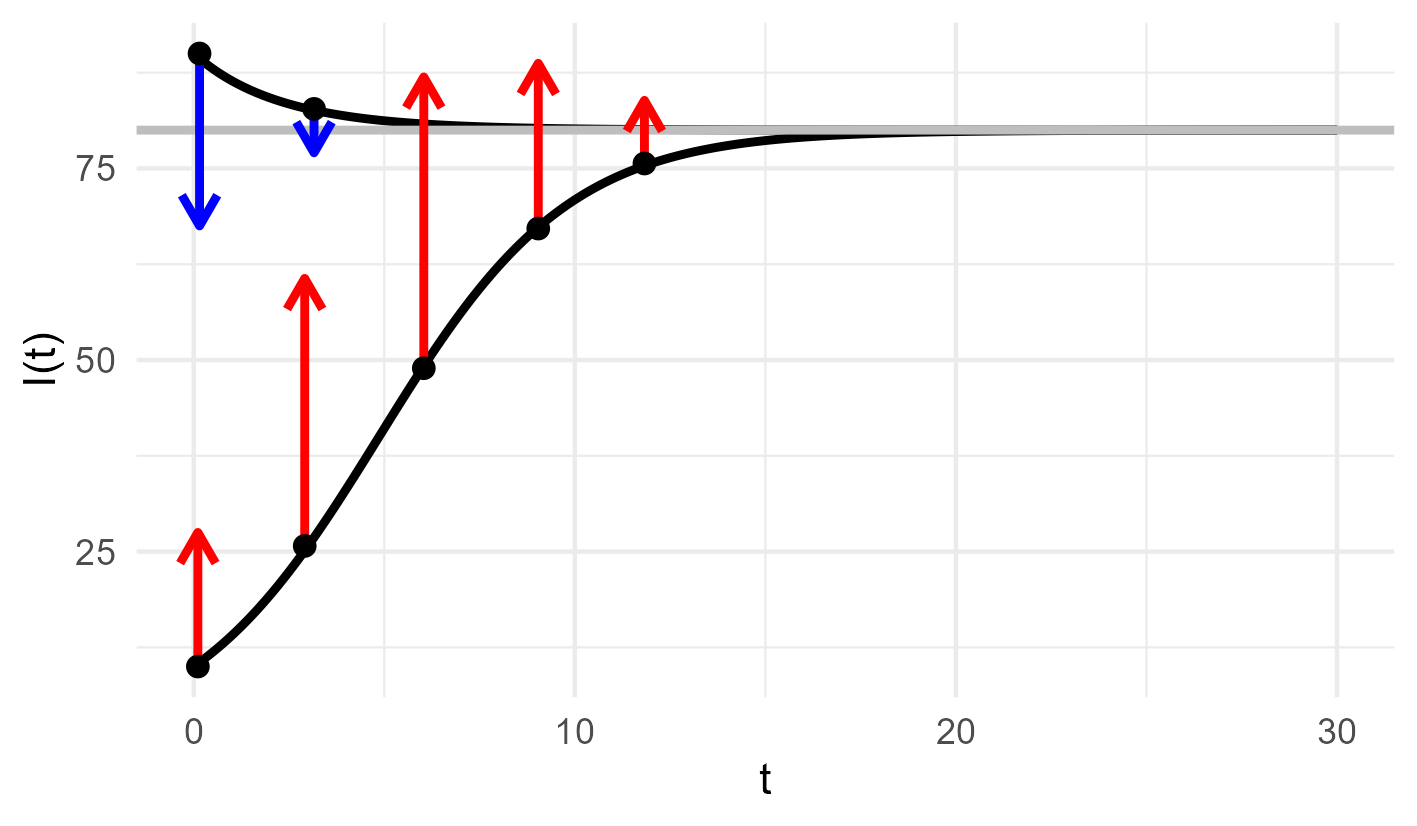
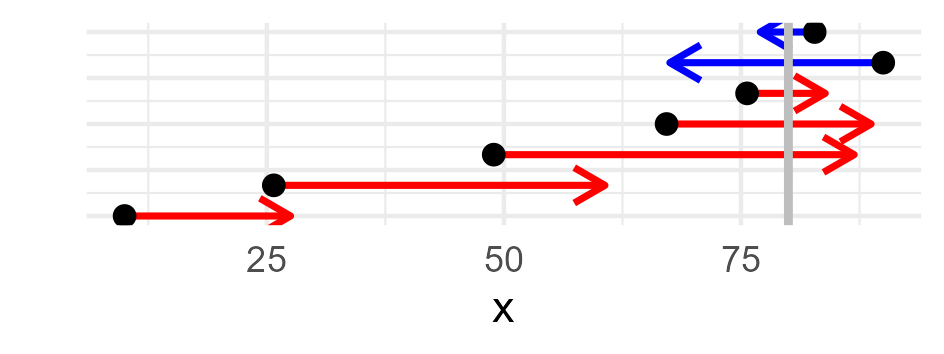
With our expert knowledge about disease transmission we were highly confident about the structure of the vector field $f$. Let’s come back to the rather opaque parameter \(\beta>0\). We admitted that we packed all the biochemical and eco-evolutionary complexities that modulate the infectiousness of the parasite into this parameter. Let’s put in some effort and make it at least time-dependent. To keep things simple, let us assume that \(t\mapsto \beta(t)\) is non-negative and periodic with fixed amplitude \(\theta_1>0\) and period \(\theta_2>0\): $\begin{equation} \beta\equiv\beta(t)\equiv\beta(t,\theta_1,\theta_2)=\theta_1\left( 1+ \sin \tfrac{2\pi t}{\theta_2}\right) \end{equation}$ Periodicity of $\beta$ makes sense for a parasite such as seasonal influenza virus from the point of view of its biochemistry. It is in dry and cold climate during winter that the protecting lipid layer becomes solid-like, the virus persists in the environment. The surface protein so important for cell entry is conserved in its polymer structure, the virus remains infectious. Not to speak of hosts crowding in trains and classrooms during cold season. Letting $\beta$ depend on time and including the amplitude and period parameters \(\theta=(\theta_1,\theta_2)\) requires some updates to the vector field $f$ of our differential equation. In its simplest form, $f$ depends only on the position $x$, but in our cases, the vector field depends also on time $t$ and \(\theta\): $\begin{equation}f_{\theta,t}(x)=(\beta(t,\theta_1,\theta_2) - \gamma) x - \tfrac{\beta(t,\theta_1,\theta_2)}{N}x^2\end{equation}$
Our differential equation know reads $\frac{dI}{dt} = f(I)$
Packing all this complexity into a sine function seems like a leap of faith bound to fail. And what is the purpose of all of this? Let’s put our model to the test of observations.
Coming back from a lengthy field trip to a remote island, your epidemiologist neighbour carries a dataset full of observations in his backpack. After some data cleaning, he gives you the number of infectious hosts recorded at \(K\) distinct time points: \(\iota(t_1),\dots,\iota(t_K)\). From his anectotal notebook about the disease you calculate the recovery rate \(\gamma\). So far, so good. In order to compare data to model outputs, we need to quantifiy how far these model outputs are away from the data by an error function: \(\begin{equation} {\cal J} = \frac{1}{K}\sum_{j=1}^{K} g(I(t_j), \iota(t_j)) \end{equation}\) To make things easier, we use \(g(x,y)=\tfrac{1}{2}|x-y|^2\) and \(J\) is called the residual sum of squares. But there is yet another way to look at ${\cal J}$. It is a functional. A functional is like a function, only that it is defined on a larger, often infinite-dimensional space such as the space of all possible solution curves to our SIS model. Remember, we keep initial conditions and \(\gamma\) fixed since we trust our epidemiologist neighbour, and the only other parameter \(\beta\) is written in a form where it depends on amplitude and period \(\theta=(\theta_1,\theta_2)\). Thus, a particular choice for \(\theta\) yields a unique solution curve \(\{I_{\theta}(t);t\geq 0\}\). The functional \({\cal J}\) maps the solution curve to a real number, the error between the model and observations.
The task of parameter inference is to find values for \((\theta_1,\theta_2)\) such that this number \({\cal J}[\{I_{\theta}(t);t\geq 0\}]\) is minimal. When minimizing functionals over a spaces of curves, we enter the calculus of variation, a mathematical field dating back to Euler and which has been properly formalized only in the last century under the name of functional analysis. Parameter inference emerged from statistics and should be considered rather a cognitive task with different computational approaches such as hypothesis testing, maximum likelihood and Bayesian updating. Whether parameter inference should actually be considered an optimization problem will spur important philosophical discussions. What is reality? The data that your epidemiologist neighbour brought back from his trip? The biochemical laws underlying disease transmission which our models seeks to mimick to the point of a caricature?
Let’s go back to the error function:
\[\begin{equation} {\cal J} = \frac{1}{K}\sum_{j=1}^{K} g(I(t_j), \iota(t_j)) \end{equation}\]If we had infinitesimal time increments of observations, we can write ${\cal J}$ as an integral:
\[\begin{equation} {\cal J} = \int_{t_1}^{t_K} g(I(t), \iota(t))dt \end{equation}\]So far, so good. There is actually a way in which the model can observe itself. Let’s revert back to our elementary model $\ref{eq:sis_simple}$, but with the addition of a control function $t\mapsto u(t)$:
$\begin{equation} \frac{dI_u}{dt} = (u(t)\beta - \gamma)I_u- \tfrac{u(t)\beta}{N}I_u^2 \end{equation}$
You can think of $u$ as a way to stir the dynamical system into a certain state. E.g. we could look for a function that will minimize the number of infections during a defined time horizon \(\begin{equation} {\cal J} = \int_{t_1}^{t_K} I_u(t) dt \end{equation}\) Instead of minimizing over \(\theta\), we would now minimize over the space of admissible control functions.
Lagrangian multiplier Hamiltonian, adjoint; Lagrangian with time-dependent multiplier Pontryagin
\[H(I, \lambda, \beta, \gamma) = L(I, t) + \lambda(t) \left[ (\beta - \gamma)I - \frac{\beta}{N}I^2 \right]\]\begin{equation} \label{eq:cauchy-schwarz} \left( \sum_{k=1}^n a_k b_k \right)^2 \leq \left( \sum_{k=1}^n a_k^2 \right) \left( \sum_{k=1}^n b_k^2 \right) \end{equation}
and by adding \label{...} inside the equation environment, we can now refer to the equation using \eqref.
Note that MathJax 3 is a major re-write of MathJax that brought a significant improvement to the loading and rendering speed, which is now on par with KaTeX.